


1. Introduction
Proteins are essential biological components and they govern essential cellular processes, from DNA replication to signaling, and their roles extend to maintaining architecture and mediating communication. Sequences determine structures, and structures determine function. Therefore, understanding the precise three-dimensional structure of proteins is crucial for predicting their functions. Traditional methods like X-ray diffraction and cryoelectron microscopy are time-consuming and expensive. However, the emergence of AlphaFold, an artificial intelligence tool, has dramatically shortened this process from years to months or even days. AlphaFold’s rapid protein structure prediction, transforming conventional methods, enables expedited exploration of these functions, offering unprecedented insights into the molecular underpinnings of life. It takes the alignment sequences of the primary amino acid sequence and homologous as inputs, uses the known structure as the training set of its deep learning, and outputs the predicted protein structural expression through the neural network model [1]. The structure prediction of protein molecules has reached the atomic level accuracy. AlphaFold holds great promise for various applications in the future, including predicting protein-ligand structures, studying allosteric pockets, protein-protein interactions, RNA targets, and designing vaccines and therapeutic proteins [2].
The advanced capabilities of AlphaFold2 have far-reaching implications for biologists and pharmacochemists. By leveraging AF2’s accurate protein structure predictions, these professionals can expedite target discovery, design potent drugs, and optimize therapeutic interventions. This groundbreaking technology holds the potential to reshape the landscape of drug development, paving the way for novel treatments and alleviating the profound health burdens imposed by conditions like cancer. HER2 is a transmembrane tyrosine kinase protein involved in cell growth, differentiation, and survival. Overexpression of HER2 is linked to several types of cancer, such as breast, stomach, and ovarian cancer. At present, there are three main types of anti-HER2 targeting drugs on the market: antibodies, ADCs and TKI. TAK-285 [3] is an investigational, small-molecule TKI. Its therapeutic potential in HER2-overexpressing brain metastasis was verified by in vivo microdialysis evaluations detecting unbound levels of TAK-285 in the extracellular space in the brain for up to 24–48h after administration. The conformational changes of HER2 before and after binding to TKI drugs analyzed by AlphaFold2 and PLIP tools can help enlighten us on potential drug development. TAK-285 has shown therapeutic potential in HER2-overexpressing brain metastasis based on in vivo microdialysis evaluations [4]. Protein-Ligand Interaction Profiler (PLIP) offers a comprehensive analysis of binding sites, hydrogen bonds, hydrophobic interactions, and other key molecular interactions, enabling researchers to unravel the mechanisms underlying protein-ligand interactions with unparalleled accuracy and detail. Using AlphaFold2 and PLIP tools to analyze the conformational changes of HER2 before and after binding to TKI drugs can provide valuable insights for potential drug development.
2. Method
2.1. Data Source for Experimental Structure.
The Protein Data Bank (PDB) [5] is a database of known crystal structures and Nuclear Magnetic Resonance (NMR) structures, many of which involve protein-ligand complexes. Currently, PDB hosts over 208,700 experimental structures and 1,068,500 Computed Structure Models. By assigning unique PDB codes to each structure, the PDB enables easy access and sharing of these critical data, facilitating scientific exploration. We collected the HER2-ligand binding structures from the Protein Data Bank (PDB) and focused on the research involving the structure with the PDB code 3RCD.The structure of HER2 Kinase Domain Complex with TAK-285 (3RCD) is analyzed by X-RAY DIFFRACTION, and TAK-285 is a novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitor. PDB format file of 3RCD was downloaded as our experimental structure manually. Since there is no experimental structure of this complex before binding, we downloaded FASTA sequences for latter prediction.
2.2. Methodology of AlphaFold2.
AlphaFold2 is an innovative protein structure prediction method developed by Google DeepMind. It has showed remarkable accuracy comparable to experimental structures in most instances and has surpassed other methods in CASP14 [6]. Its principle is rooted in deep learning techniques and computer vision, simulating physical principles to predict protein structures. AlphaFold2 serves as a system that predicts the 3D structure of an input protein through its sequence. To achieve this, it utilizes various protein databases and open-source programs.
The pivotal components within AlphaFold2, namely Evoformer and Structure module, harness the multiple sequence alignment (MSA) representation and pair representation as inputs, thereby generating the anticipated 3D structure. In the process, AlphaFold2 effectively employs the HMMER software to locate homologous sequences within the Uniprot and MGnify sequence databases. Utilizing HMMER, it constructs and furnishes the MSA of the provided protein sequence along with its identified homologues. Furthermore, AlphaFold2 integrates the HH-suite package to assess the availability of 3D structures of related proteins in the Protein Data Bank (PDB). This comprehensive approach ensures a robust and multi-faceted prediction of protein structures.
2.3. Data Source for Computed Structure.
To obtain predicted models of the HER2 Kinase Domain before binding to TAK-285, we accessed AlphaFold2 through Google Colaboratory [7]. We input the downloaded protein FASTA sequences, and set msa_mode as mmseqs2_uniref_env, num_recycles as 6 to generate the top 5 predicted models.
2.4. Method of Alignment.
We imported the experimental and computed structures into Pymol. The alignment fuction of Pymol enabled us to visualize the conformational changes of HER2 before and after binding to TAK-285 effectively.
3. Results and Discussion
3.1. Evaluation of Predicted Models.
The assessment of the top 5 predicted models based on the pLDDT metric reveals encouraging findings, as all models achieve scores above 80, which are deemed to be optimal. The pLDDT values for the top 5 model are: 84.7, 84, 83.1, 82.1, 82. Consequently, the model scores the highest had been chosen for further alignment and analysis. The robust pLDDT scores signify the precision and credibility of the predicted structures, underscoring the effectiveness of our method in forecasting the HER2 Kinase Domain’s conformation. These promising outcomes pave the way for valuable insights into the interaction between HER2 and TAK-285, offering significant potential in guiding drug development approaches for HER2-overexpressing cancers.
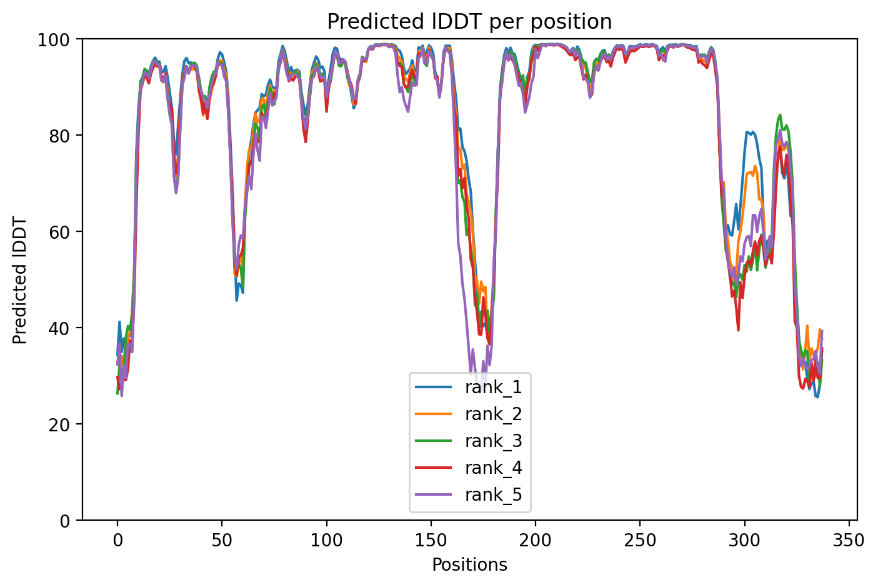
Figure 1. Predicted IDDT per position.
As observed from figure 1, a few wave cliffs are noticeable, with their positions primarily clustered around 0-10A, 60-70A, 170-180A, 290-300A, and 325-336A. These specific positions are predominantly indicative of the protein’s looser regions.
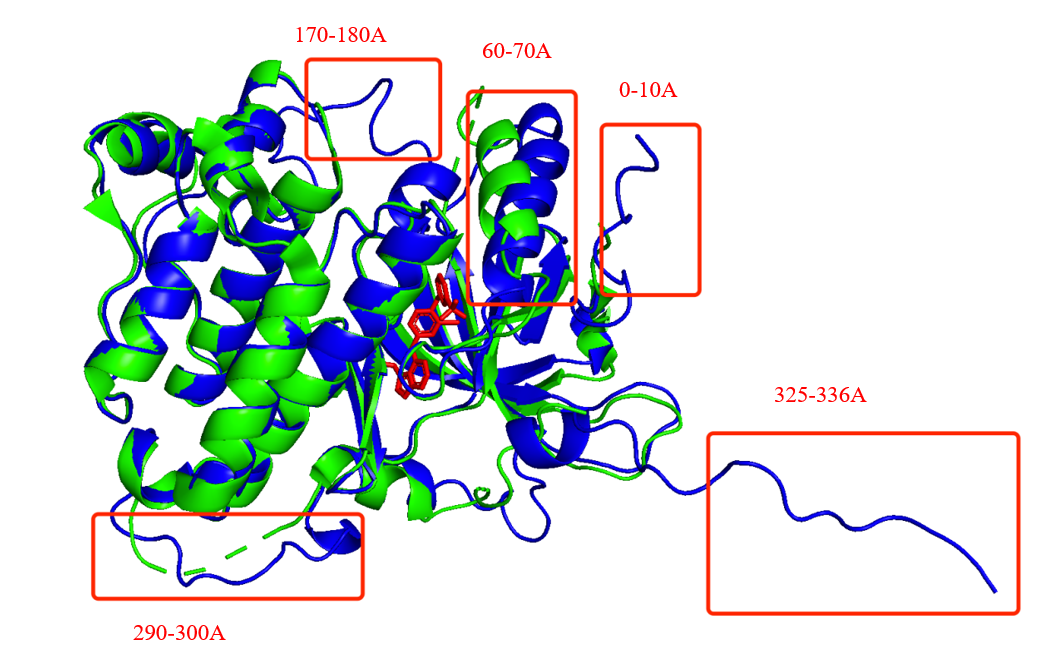
AlphaFold Experiment TAK-285
Figure 2. Significant differences between the experimental and computed structures.
Interestingly, within the range of 60-70A, a significant alteration in the structure of an α-helix is observed. Several factors could account for this change. One possibility is the potential inaccuracy of AlphaFold’s prediction, as indicated by the pLDDT score of 50 in that region. Moreover, the proximity of the spiral’s tail to the small molecule implies that the insertion of TAK-285, along with its associated weight, might induce alterations in van der Waals forces, leading to conformational changes in the surrounding environment and subsequently affecting the α-helix’s conformation. Another possible explanation could be the presence of a covalent bond between TAK-285 and the α-helix.
3.2. Pymol Alignment Results.
Analyzing the right half of Figure 2 reveals that several α-helices distant from the ligand remain unchanged before and after binding. However, the rest of the alignment shows an obvious structural difference, particularly in the random coils. These observations support the idea that the general conformation of HER2 would not change and would only induce local changes due to the binding.
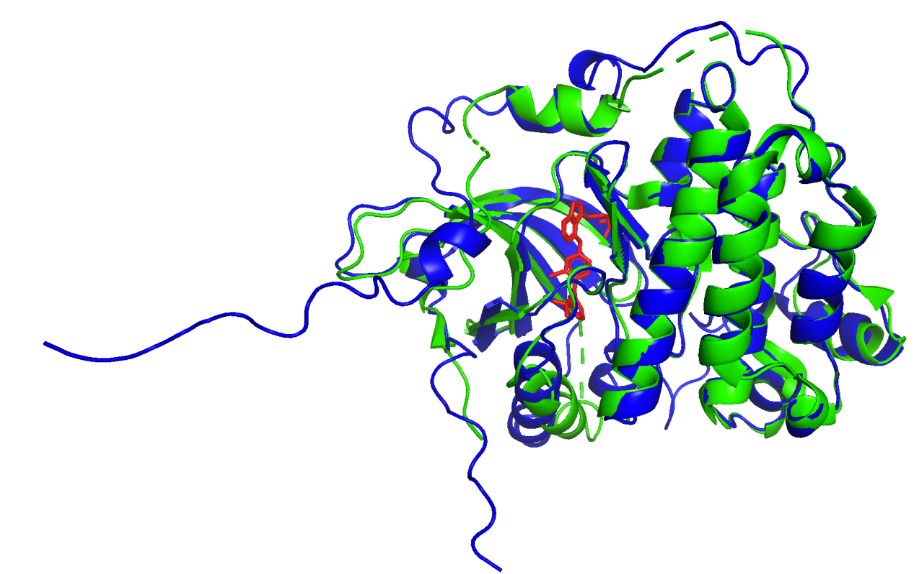
AlphaFold Experiment TAK-285
Figure 3. Pymol alignment results (AlphaFold Experiment TAK-285).
3.3. Binding Sites in 3RCD.
The protein-ligand interaction outcomes presented below were acquired using PLIP [8], a robust tool designed for the analysis of such interactions. The findings from PLIP unveil the intricate network of interactions between the HER2 Kinase Domain and TAK-285, emphasizing key regions crucial for ligand binding. These binding sites play a pivotal role in the modulation of the protein’s function and hold potential as targets for therapeutic interventions.
|
|
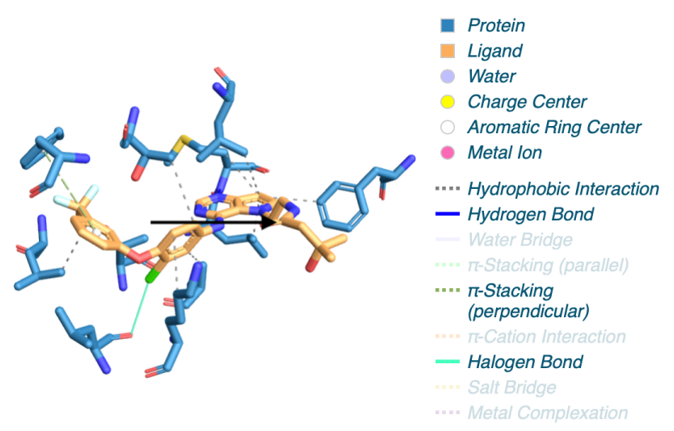
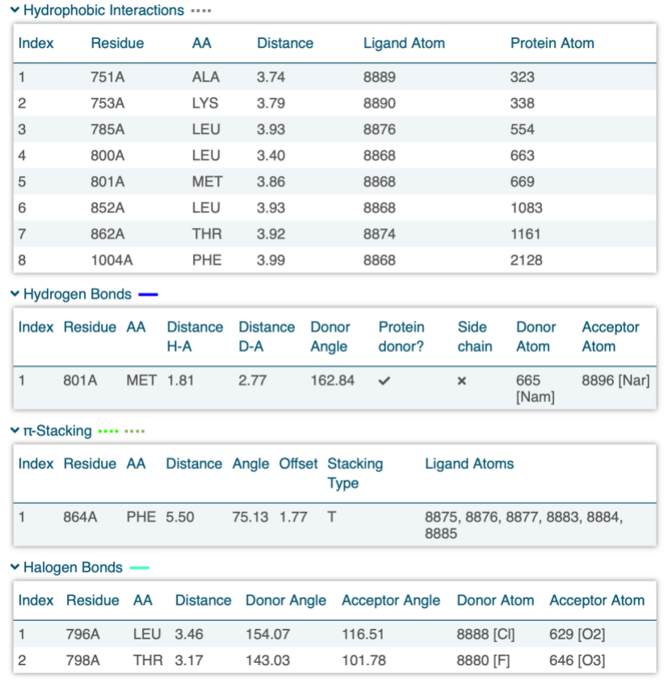
Figure 4. Protein-ligand interaction result of 3RCD.
As discussed in section 3.1, the conformational change of the α-helix (60-70A) could potentially arise from a covalent bond. However, the corresponding protein-ligand interaction is not listed above. This absence might be due to the lack of an interaction in that region or an interaction that was not displayed in the list due to PLIP’s settings. PLIP’s default parameters set a threshold, and these unchanged settings could result in the covalent bond not being recognized by PLIP. Exploring this region could also provide a viable avenue for drug discovery research.
To visualize the structural changes, we listed the 3D structures of those binding sites:
(a)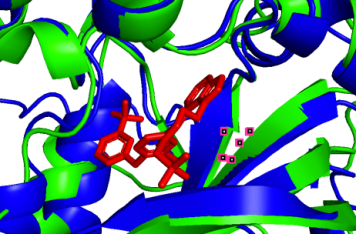 (b)
(b) 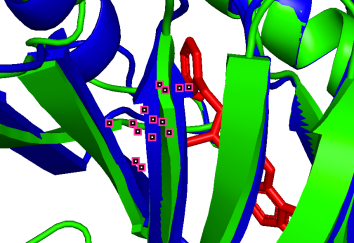 (c)
(c) 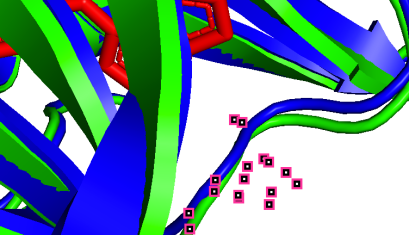
751A-50A (pLDDT=90) 753A-52A (pLDDT=90) 800A-99A (pLDDT=90)
(d) 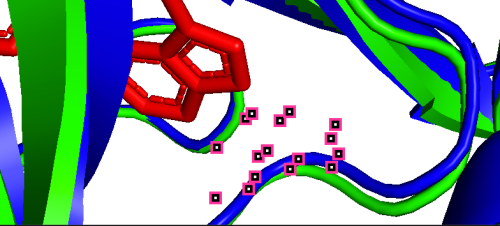 (e)
(e) 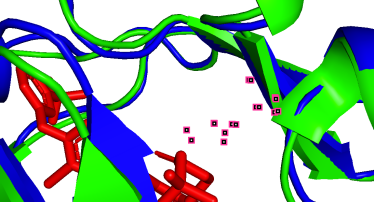 (f)
(f) 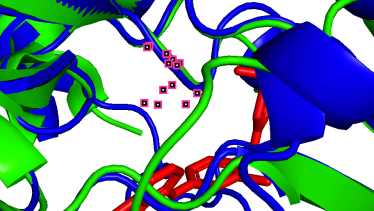
801A-100A (pLDDT=90) 852A-151A (pLDDT=98) 862A-161A (pLDDT=98)
(g) 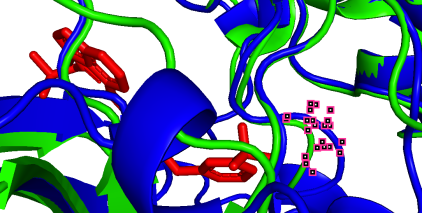 (h)
(h) 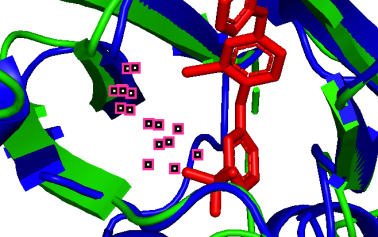 (i)
(i) 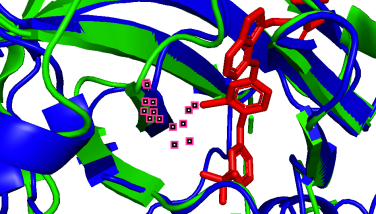
864A-163A (pLDDT=90) 796A-95A (pLDDT=90) 798A-97A (pLDDT=90)
(j) 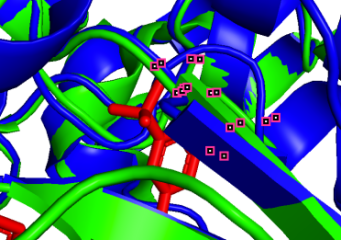 (k)
(k) 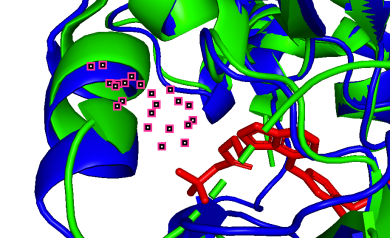
785A-84A (pLDDT=80) 1004A-303A (pLDDT=70)
Figure 5. Visualized conformational changes of 3RCD.
As depicted in the figure above, we observe three types of situations for the binding sites:
(a) Precise prediction with minimal conformational changes: Hydrophobic interactions are present, but the extent of conformational changes is limited, which possibly due to TAK-285 being a small molecule. Another possibility is the inherent flexibility of the protein structure, where the slight changes after binding align with the protein’s motion before binding, as detected by X-ray diffraction.
(b-i) Precise prediction with certain conformational changes: Half of the locations (c, d, f, g) consist of random coils, which easily change their structures due to interactions. The other locations (b, e, h, i) are not random coils but still experience structural changes, likely induced by non-covalent bonds.
(j, k) Less precise prediction with certain conformational changes: In this scenario, changes may result from interactions between TAK-285 and the protein or possible inaccuracies in the model predictions.
Analyzing these different binding site situations provides valuable insights into the intricate protein-ligand interactions and their impact on HER2’s conformational changes upon binding to TAK-285. These findings advance our understanding of the molecular mechanisms underlying the HER2-TAK-285 complex and offer critical information for potential drug design and targeted therapeutic strategies. By deciphering the binding sites and their interactions, our study gains deeper understanding of the molecular mechanisms governing HER2’s response to TAK-285. This knowledge has significant implications for drug design and development, facilitating the identification of key binding pockets that may be utilized in designing novel therapeutic agents for HER2-related cancers. Moreover, it contributes to the broader field of structure-based drug discovery, opening avenues for more precise and effective treatments in the future.
4. Conclusion
This study successfully demonstrates that ligands can induce structural changes in binding sites. Analyzing and visualizing these changes provides valuable insights for drug discovery. While AlphaFold2 represents a significant advancement in protein structure prediction, it does exhibit certain limitations. For example, the prediction of multi-chain protein complexes remains challenging, the effect of mutation on the structures cannot be predicted, the many possible conformational states of a protein, such as static, dynamic, monomer, complex, etc., cannot be accurately captured and distinguished, scientists and researchers are already using AlphaFold for drug development. In the future, enhancing AlphaFold2’s ability to predict structures of greater complexity, addressing rare or unconventional protein conformations, and improving the consideration of post-translational modifications will likely be key directions for its continued development and optimization. In late January 2020, DeepMind scientists used AlphaFold2 to map the SARS-COV-2 virus’s protein structure [9] - which was later experimentally confirmed to be accurate. Subsequently, virologists around the world began using AlphaFold2 to study the novel coronavirus. And researchers are utilizing AlphaFold and related tools to interpret experimental data generated using X-ray crystallography and cryo-electron microscopy. An example is Marcelo Sousa, a biochemist at the University of Colorado Boulder, who employed AlphaFold to make models from X-ray data of proteins that bacteria use to evade an antibiotic called colistin [10]. To address the challenge of predicting multi-chain protein complexes accurately, DeepMind introduced an additional AlphaFold model designed for multimeric inputs with established stoichiometry, named AlphaFold-Multimer [11]. This model substantially improves the precision of predicted multimeric interfaces compared to the input-adapted single-chain AlphaFold, all while retaining a high level of accuracy within individual chains.
Structure-based drug discovery (SBDD) has long been a key approach for identifying hit molecules and optimizing leads. AlphaFold’s ability to predict protein structures makes it a powerful tool for identifying hits for novel targets with limited or no existing structure information [12]. By leveraging the potential of AlphaFold and similar tools, researchers can enhance their understanding of protein-ligand interactions and streamline the drug discovery process. The insights gained from such studies hold immense promise for advancing drug development and precision medicine, enabling more effective and targeted therapeutic interventions.