


1. Introduction
Sunitinib is an orally administered, low molecular weight compound that functions as a multi-targeted inhibitor of receptor tyrosine kinases. This pharmaceutical agent was produced by Pfizer. The compound in question is a derivative of indole and pyrrole, possessing a molecular weight of 398.47. It exists as an orange solid under standard room temperature conditions. Additionally, it demonstrates a partition coefficient between oil and water of 3.1, a dissociation constant of 8.5, and also known as N-[2-(diethylamino)ethyl]-5-[(Z)-5-fluoro-1,2-dihydro-2-oxo-3H-indol-3-ylmethylene]-2,4-dimethyl-1H-pyrrole-3-formamide in the field of chemistry. The chemical under investigation exhibits a dual functionality as a multi-receptor tyrosine kinase inhibitor and a highly effective ATP-competitive type I1/2B inhibitor of cell cycle protein-dependent kinase 2 [1]. Polar interactions are formed with Glutamic acid 81, Leucine 83, and Aspartic acid 86 inside the hinge region of cell cycle protein-dependent kinase 2 [1]. In 2006, the Food and Drug Administration (FDA) granted permission to Sunitinib for its utilization in the management of gastrointestinal mesenchymal tumor (GIST) and advanced or metastatic renal cell carcinoma (RCC) that demonstrate resistance to imatinib. Furthermore, in the year 2011, approval was granted for the utilization of this medication in managing metastatic highly differentiated progressive pancreatic neuroendocrine tumors [2]. Sunitinib is frequently employed for the treatment of GIST and RCC, as it is regularly prescribed for these specific medical conditions. The primary aim of this research is to examine the synthetic pathway utilized for the production of Sunitinib, as well as its potential utilization in the treatment of GIST and RCC.
2. GIST
GIST is presently the prevailing mesenchymal neoplasm, and its etiology is characterized by the prevalence of mutations in the gene function of C-KIT or PDGFR α, with a minority of cases exhibiting additional molecular alterations such as mutations in the genes of SDHX, BRAF, NF1, K/N-RAS, and PIK3CA, among others. The yearly occurrence rate of GIST is estimated to be approximately 10-15 million instances globally [3]. China has an estimated annual incidence of 20,000-30,000 new cases, whereas the United States witnesses over 5,000 newly diagnosed individuals on a yearly basis [3]. The stomach was identified as the predominant location of origin, accounting for 55.6 percent of cases. This was followed by the small intestine at 31.8 percent, colorectum at 6.0 percent, oesophagus at 0.7 percent, and other sites at 5.5 percent [3]. GIST is usually asymptomatic and is usually an incidental finding on imaging. However, clinical manifestations may include abdominal pain or gastrointestinal bleeding. Imatinib can be used to treat GIST, but a large percentage of patients develop resistance. In contrast, sunitinib and its malate (sunitinib malate) can be used for second-line treatment of GIST that are resistant to imatinib. Its mechanism of action is to block receptor tyrosine kinase signaling through all three isoforms of KIT, PDGFRs and VEGFR. VEGF, which is unique to sunitinib malate, inhibits tumour-associated angiogenesis [4]. The administration of Sunitinib has exhibited substantial effectiveness in improving the duration of tumor progression, survival without progression, and overall survival in patients with advanced GIST who have previously failed treatment with another TKI, as compared to the administration of an inactive substance (placebo) [4].Treatment with sunitinib is usually well tolerated and rarely leads to discontinuation of therapy. And relevant case reports have shown no adverse surgical events with surgical sunitinib [4].
3. RCC
RCC is a prevalent and highly lethal malignant neoplasm affecting the kidneys and is characterized by a significant propensity for morbidity and mortality [5]. The aforementioned subtype of kidney cancer, which constitutes over 90% of all occurrences, is derived from renal tubular epithelial cells [6]. The incidence of kidney cancer is higher in populations residing in Europe and North America, while it is comparatively lower in Asia. In the year 2013, RCC was acknowledged as the sixth most prevalent form of cancer, resulting in an annual mortality rate above 140,000 individuals. RCC constitutes around 2-3% of the total incidence of malignant neoplasms. It is recognized as the most lethal genitourinary malignancy, with a fatality rate ranging from 30 to 40 percent. In contrast, bladder and prostate malignancies demonstrate comparatively lower mortality rates, approximately 20 percent. The prevalence of this phenomenon is on the rise, with a larger occurrence observed in industrialized nations compared to developing nations. Furthermore, it is noteworthy to mention that the incidence of this illness exhibits a higher occurrence among boys in comparison to females, with a male to female ratio of 1.5:1 [7]. Additionally, the mortality rate is observed to be higher in males as compared to females [7]. RCC is believed to arise as a result of a process that induces mutations within the VEGFR pathway [8]. In the past decade, Sunitinib has emerged as the primary targeted therapeutic agent for the treatment of RCC. Its mechanism of actiormalities and the induction of vascular normalisation, so effectively addressing the disease. Nevertheless, it is commonly observed that a majority of individuals exhibit resistance to sunitinib treatment within a timeframe ranging from 6 to 15 months [9].
4. Actions of Sunitinib
Sunitinib's drug activity is that it can inhibit protein kinases, antagonise or inhibit the development of new blood vessels and inhibit or prevent tumour proliferation. Its mechanism of action is to terminate the anti-angiogenesis mechanism that supplies blood to tumour cells and the mechanism of direct attack on tumour cells. However, it also has certain toxic side effects. Sunitinib has some hepatotoxicity. Several cases have been reported in which treatment resulted in clinically significant liver damage, with onset occurring after several cycles of treatment [10]. It also causes hyperammonaemia in rare cancer patients treated with conventional or even low oral doses, manifested by confusion and irritability, mild elevations of serum enzymes and bilirubin, and significant elevations of serum ammonia [10]. The toxicity profile of the aforementioned combination treatment is as follows: A 57-year-old female patient initiated sunitinib therapy due to the recurrence of metastatic gastrointestinal mesenchymal stromal tumor following the ineffectiveness of imatinib. The patient's condition remained stable, and her liver function remained within normal parameters throughout eight cycles of sunitinib treatment, which consisted of four weeks of administration followed by a two-week discontinuation period. The patient's current pharmacological regimen consisted of acetaminophen, in addition to conventional asthma drugs. In the eighth cycle, the patient was administered oral levothyroxine as a means of managing hypothyroidism prior to commencing sunitinib treatment. Subsequently, she was admitted to the hospital due to a gradual increase in circulating liver enzyme levels. Despite the cessation of sunitinib treatment and the implementation of a rigorous regimen of supportive therapy, the patient's demise occurred four days subsequent to her hospital admission. During the postmortem examination, the liver exhibited pronounced centrilobular necrosis accompanied by moderate to severe steatosis and a minor infiltration of the tumor inside the parenchyma. The results of the viral staining assay yielded a negative outcome. The incidence of liver failure in patients undergoing sunitinib treatment is infrequent. The autopsy results excluded the possibility of tumor disease development and viral infection as causes of death. The patient's demise is plausibly attributed to the synergistic interplay between sunitinib, acetaminophen, and levothyroxine [11]. The specific mechanism through which the combination leads to liver failure remains unknown.
Not only that, but sunitinib may also have some toxic side effects in pregnant women and infants. Since blood vessel formation is critical to embryonic and fetal development, and sunitinib inhibits blood vessel formation may have adverse effects on pregnancy, although it has not been adequately studied in pregnant women in a rigorously controlled manner. Hence, it is crucial to guarantee that patients receive sufficient education regarding the potential hazards linked to the use of sunitinib on the developing fetus during pregnancy or in the case of pregnancy coinciding with sunitinib treatment. Additionally, it is advisable to counsel women who are capable of carrying children to employ contraceptive measures while undergoing sunitinib therapy. Empirical evidence has been shown to substantiate the excretion of sunitinib and its metabolites in the lactation process of rats. The study observed that the levels of sunitinib and its metabolites in the milk of lactating female rats were found to be up to 12 times more than their levels in the plasma following administration of a dosage of 15 mg/kg. The lack of clarity regarding the elimination of sunitinib and its key active metabolite in human breast milk remains unresolved. Given the regular excretion of pharmaceutical substances in human breast milk and the potential for substantial adverse effects on infants, it is imperative to thoroughly deliberate the decision between ceasing breastfeeding or terminating medicine in lactating mothers. This decision should be made by considering the significance of the drug to the mother during her course of treatment.
The pharmacokinetics of drugs exhibit comparable characteristics in both healthy individuals and patient populations undergoing testing, namely those diagnosed with solid tumors such as GIST and RCC. The process of sunitinib absorption in the human body typically results in peak plasma concentrations occurring within a time frame of 6 to 12 hours subsequent to oral administration of a single dose. The concurrent administration of meals and sunitinib did not have a significant impact on its bioavailability. The principal metabolic pathway of sunitinib is mediated by the cytochrome P450 enzyme CYP3A4, resulting in the formation of its primary active metabolite. Following this, the specific metabolite undergoes additional metabolic changes assisted by the enzyme CYP3A4. The observed terminal half-lives of sunitinib and its primary active metabolite have been demonstrated to vary between 40 and 60 hours and 80 and 110 hours, respectively. After many administrations, sunitinib reveals a notable rise in accumulation inside the body, with a 3-4-fold increase observed. Similarly, its major metabolite exhibits a more substantial increase in accumulation, with a 7-10-fold increase observed. These accumulations lead to the attainment of steady-state concentrations within a span of 10-14 days. The primary route of metabolism for this substance takes place outside of the body, primarily by fecal excretion. Approximately 61% of the administered dose is eliminated through feces, while renal excretion of the medication and its metabolites accounts for approximately 16% of the dose.
5. Synthetic route
5.1. Synthetic route 1
Sunitinib (1) was synthesised from 5-fluoro-1,3-dihydroindol-2-one (2) by a two-step process of Knoevenagel condensation followed by amidation reaction as shown in Figure 1. The synthesis of Sunitinib (1) was carried out using 5-fluoro-1,3-dihydroindol-2-one (2). The intermediate product (5) was synthesized using the Knoevenagel condensation reaction between 5-fluoro-1,3-dihydroindol-2-one (2) and 5-formyl-2,4-dimethyl-1-H-pyrrole-3-carboxylic acid (4). Subsequently, compound (5) underwent amidation with N,N-diethylethylenediamine (3) to yield the desired chemical sunitinib (1) [12]. The key to this synthetic route lies in the stereoselective construction of (Z)-type double bonds in the molecular structure via the Knoevenagel condensation reaction [12].
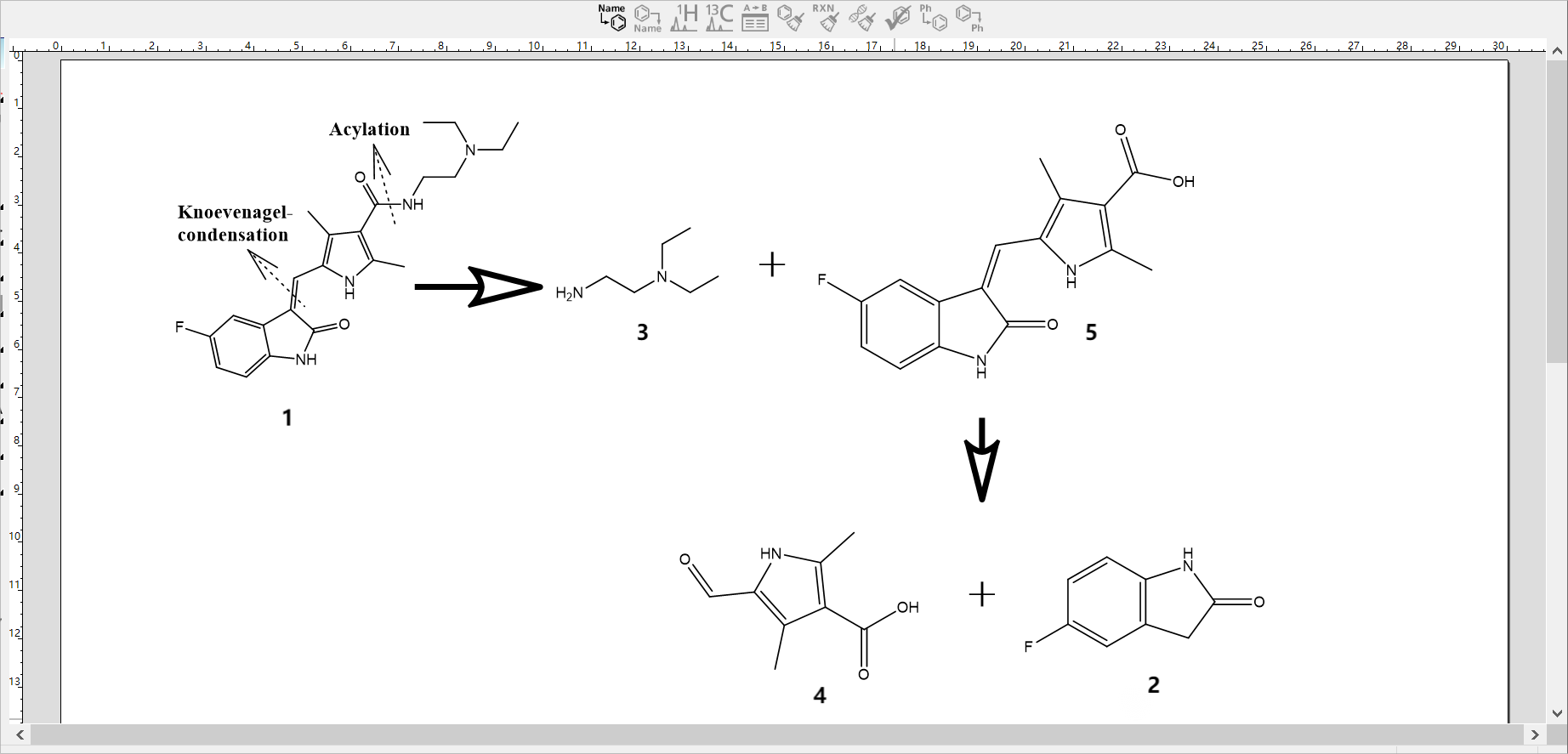
Figure 1. Synthetic route 1 [12].
5.2. Synthetic route 2
The synthetic route depicted in Figure 2 involved the utilization of 4-fluoro-2-iodoaniline (7) as a starting material. Through the process of chloroacetylation, 2-chloro-N-(4-fluoro-2-iodo-phenyl)-acetamide (8) was obtained. Subsequently, 8 was subjected to a reaction with triethyl phosphite, resulting in the formation of diethyl [(4-fluoro-2-iodo-phenylcarbamoyl)-methyl] phosphonate (9) under solvent-free conditions. Furthermore, compounds 9 and 6 were subjected to a condensation reaction under the presence of potassium hydroxide, leading to the formation of compound (10). Finally, in the presence of palladium acetate and triethylamine, the desired compound (1) was obtained through a one-step intramolecular 5-exo type cycloaddition reaction, which facilitated the construction of the indolone and Z-bond structure [13].
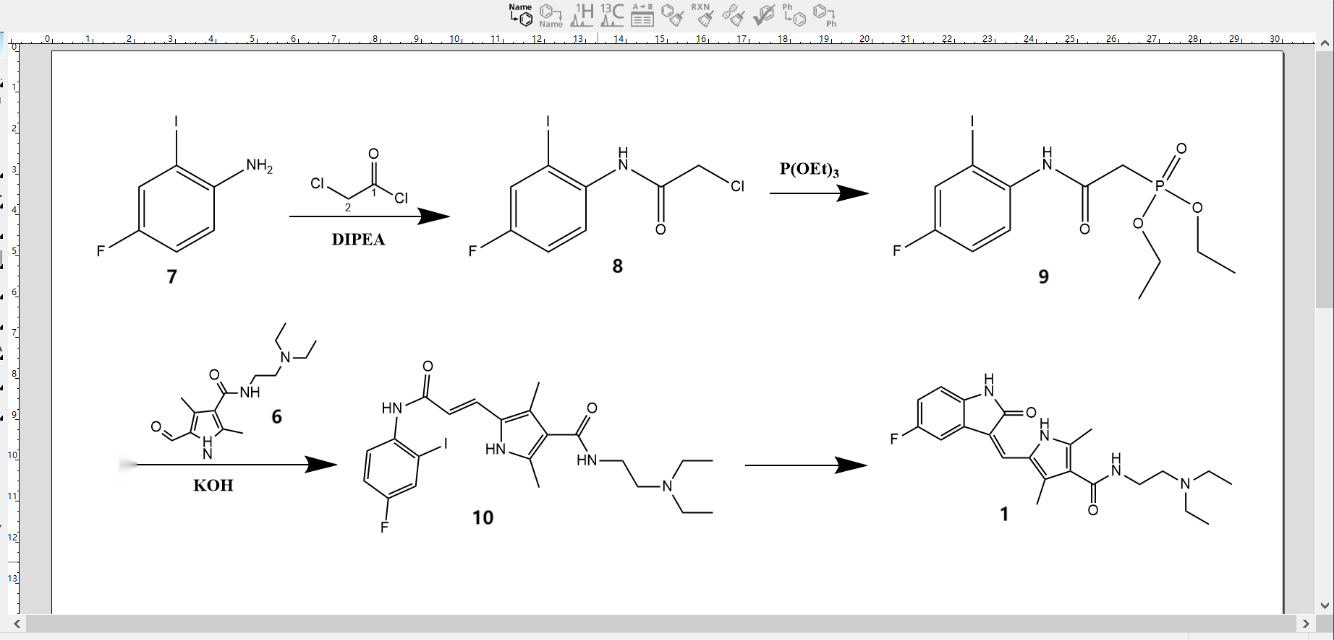
Figure 2. Synthetic route 2 [13].
5.3. Synthetic route 3
Figure 3 depicts the synthetic pathway 3, which outlines the process of synthesizing diethyl 3,5-dimethyl-1H-pyrrole-2,4-dicarboxylate (12) using the oximation and reduction one-pot (Knorr) technique. This synthesis commences with the utilization of ethyl acetoacetate (11). The synthesis of ethyl 2,4-dimethyl-1H-pyrrole-3-carboxylate (14) was achieved by decarboxylation through hydrolysis.Compound 14 was produced through the use of the Vilsmeier-Haack formylation reaction, followed by a process of hydrolysis and subsequent condensation with N-diethylethylenediamine. The molecule identified as N-(2-(diethylamino)ethyl)-2,4-dimethyl-1H-pyrrole-3-carboxamide was obtained by the utilization of a synthetic process (17). In addition, p-fluoroaniline (18), chloral hydrate, and hydroxylamine hydrochloride were used as raw materials, which were first acylated and oxidised, while 5-fluoroindigo (20) was obtained by cyclisation in concentrated sulphuric acid, and 5-fluoro-1,3-dihydroindol-2-one (2) was obtained by reduction with flavomelon. Finally, the condensation of the 3-position active methylene group of 2 with the 5-position formyl group of 17 was utilised in the presence of triethylamine to give (1) [14].
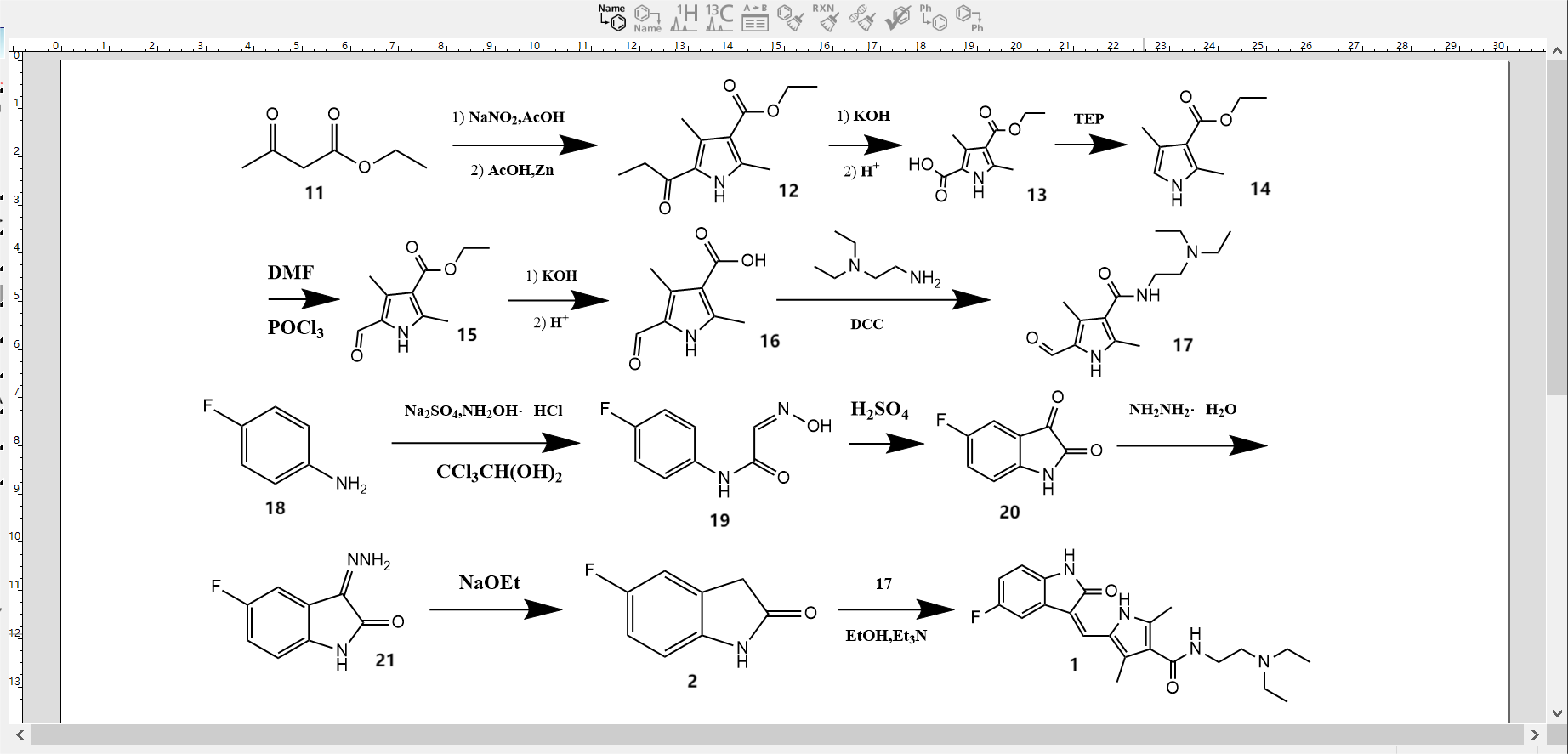
Figure 3. Synthetic route 3 [14].
5.4. Evaluation and summary
Synthesis route 1 is based on 5-fluoro-1,3-dihydroindol-2-one (2) as raw material, but in the process of raw material preparation of waste acid and the generation of dark-coloured by-products post-treatment is difficult, and environmental pollution; and hydrazine hydrate reduction reaction (Wolff-Kishner reaction) needs to be used in excess. Alternatively, a substantial surplus of hydrazine hydrate can be employed to enhance the reaction's output, but with inherent hazards associated with the manufacturing procedure. Therefore, it is recommended that the synthesis of 5-fluoro-1,3-dihydroindol-2-one (2) should be avoided as much as possible [12].
The synthetic route 2 is characterised by the construction of a p-fluoroaromatic ring in place of the pyrrole heterocyclic skeleton, followed by the formation of the indolone ring. The synthesis yielded a sample with HPLC purity of 99.4%, with only one single impurity greater than 0.1%; this impurity is presumed to be a double-bonded stereoisomer of sunitinib based on HPLC-MS test results [12]. Avoiding the patent protection of foreign processes while having a high yield of about 75%, it can be used in modern industrial production [13].
Synthetic route 3 had readily available raw materials, easy to manipulate reaction conditions, and the intermediates in each step were solid and easy to purify, but the final overall yield was very low at 13.2% [14]. Furthermore, an excessive amount of diamine was employed during the amidation reaction, leading to difficulties in the following elimination of the surplus amine. Furthermore, a limited number of amide coupling reagents, such as EDC and CDI, yielded satisfactory outcomes. Additionally, the elimination of by-products generated by the activator during these coupling reactions posed a significant problem [15].
In summary, comparing the above three synthetic routes, the synthetic route 2 is safer and more effective, easy to operate, and green and environmentally friendly, which is suitable for modern industrial production.
6. Conclusion
Sunitinib is a class of compounds that block the action of tyrosine kinases and is a first-generation TKI. This pharmacological drug is prescribed for the therapeutic treatment of GIST, advanced RCC, and metastatic pancreatic neuroendocrine tumors with high differentiation that have demonstrated resistance to imatinib. In this paper, we study the pharmacological activity and toxicity of sunitinib, the incidence and pathogenesis of the diseases targeted for treatment and the regulatory mechanisms involved in its treatment of the diseases, and then analyse and compare the synthetic pathways of sunitinib developed by three previous researchers, and finally propose an evaluation and recommendation of the synthetic pathways. Sunitinib has been the subject of substantial clinical research in the treatment of GIST and advanced RCC that have demonstrated resistance to imatinib. Numerous researchers have conducted experiments to investigate the relevant aspects. The topics addressed in this paper are of utmost importance for the advancement of industrial production of sunitinib and the future therapeutic approaches for patients with gastrointestinal mesenchymal tumors and advanced renal cell carcinoma resistant to imatinib. Future studies should focus on the mechanism of action behind the toxicity of sunitinib in combination with other drugs, investigate a more optimal synthesis pathway for sunitinib, and further explore whether other drugs in combination with sunitinib can improve the efficacy of the drug and reduce the drug's tolerability in the treatment of cancer tumours.